|
“X-連鎖MAGT1缺陷伴随EBV易感性增加及N-連接糖基化缺陷”(XMEN)疾病是一種(zhǒng)罕見的聯合免疫缺陷疾病(CID),由鎂離子轉運蛋白1 (MAGT1)基因功能(néng)缺失突變所引起(qǐ)[1, 2]。MAGT1的功能(néng)缺陷會(huì)損害鎂離子轉運及系列蛋白的N-糖基化,從而導緻多個關鍵免疫受體(如NKG2D)表達缺失,繼而誘發(fā)免疫系統異常、慢性EBV感染和腫瘤形成(chéng)[1, 2]。此外,非病毒性肝酶異常升高也是XMEN患者的主要病征。目前尚無針對(duì)XMEN疾病的特定有效治療方法。XMEN患者存在反複感染,生存質量較差,大部分患者20-40歲死亡[2]。
2023年4月21日,複旦大學(xué)附屬兒科醫院/生物醫學(xué)研究院羅敏/盧智剛教授團隊在Journal of Allergy and Clinical Immunology在線發(fā)表了Epigenetic activation of the TUSC3 gene as a potential therapy for XMEN disease論文。患者的主診醫生複旦大學(xué)附屬兒科醫院臨床免疫科侯佳副主任醫生共同指導參與該項研究。

2020年12月臨床免疫科侯佳醫生在門診接診了一對(duì)來自江西的兄弟,自幼經(jīng)常感染生病,母親遞上厚厚一沓病例資料,滿面(miàn)愁容不知所措。基因報告顯示兄弟倆有一個MAGT1基因突變位點,這(zhè)是聯合免疫缺陷病的緻病基因之一,極爲罕見。在做了全面(miàn)臨床評估後(hòu),判斷兄弟倆的反複肺炎、EBV感染和肝功能(néng)異常符合MAGT1突變所緻XMEN疾病的臨床特征。然而在查閱大量文獻後(hòu),卻發(fā)現國(guó)際上對(duì)XMEN疾病尚無有效的治療方案。這(zhè)一結果讓主診醫生有種(zhǒng)無力感,當時(shí)給到的方案隻能(néng)是控制感染、對(duì)症治療和随訪觀察。在一次學(xué)術交流中,侯佳醫生與羅敏研究員提到這(zhè)兩(liǎng)個一直牽挂于心的患者,希望能(néng)從MAGT1基因功能(néng)和機制上尋找到治療的突破口。曆時(shí)2年多,在臨床和科研團隊的不懈努力下,終于有了重要發(fā)現,并篩選到很有治療前景的表觀遺傳藥物。
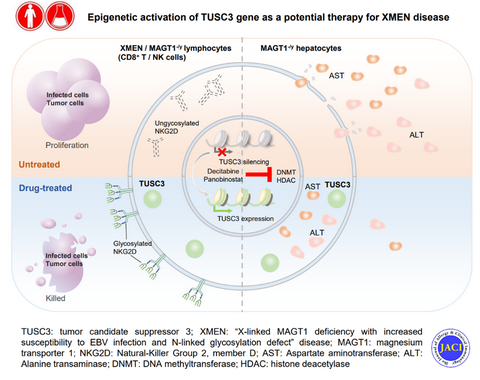
MAGT1和TUSC3在鎂離子轉運和蛋白N-糖基化功能(néng)上表現出功能(néng)冗餘[3-6]。研究首先利用組織樣本及多個數據庫分析了MAGT1和TUSC3的人體組織表達譜,并系統分析其與XMEN患者病征的相關性。結果顯示TUSC3雖然在多種(zhǒng)器官組織中廣譜表達,但是在免疫系統和肝髒中并不表達,與XMEN患者的主要病變組織一緻;也提示重新激活TUSC3的表達有可能(néng)恢複XMEN免疫細胞的功能(néng)。
鑒于XMEN是一種(zhǒng)極罕見的遺傳疾病,目前XMEN疾病相關的研究模型非常有限。患者來源的細胞極其稀少、細胞增殖數量有限;而小鼠模型與人的表型存在一定差異。研究利用人來源的NKL細胞構建了MAGT1 KO細胞系,成(chéng)功模拟了XMEN患者來源淋巴細胞的缺陷表型,爲研究XMEN疾病的發(fā)病機制和治療策略提供了可靠的細胞模型平台。
基于以上細胞模型,作者證明TUSC3能(néng)夠顯著恢複MAGT1 KO 所導緻的免疫缺陷。研究繼而利用該模型進(jìn)行小分子藥物篩選,鑒定出兩(liǎng)種(zhǒng)表觀調控藥物:地西他濱和帕比司他,能(néng)夠顯著上調 TUSC3表達,并修複XMEN免疫缺陷和肝酶異常;藥物的治療效果在XMEN患者來源的免疫細胞上得到了進(jìn)一步的證明。該研究爲XMEN疾病的有效治療提供了新的策略。
複旦大學(xué)附屬兒科醫院碩士丁浩東爲該文的第一作者;複旦大學(xué)附屬兒科醫院/生物醫學(xué)研究院羅敏研究員、生物醫學(xué)研究院盧智剛研究員,以及複旦大學(xué)附屬兒科醫院免疫科侯佳副主任醫師爲該論文的共同通訊作者。
原文鏈接:https://doi.org/10.1016/j.jaci.2023.04.003
參考文獻
[1] CHAIGNE-DELALANDE B, LI F Y, O'CONNOR G M, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D [J]. Science, 2013, 341(6142): 186-91.
[2] RAVELL J C, CHAUVIN S D, HE T, et al. An Update on XMEN Disease [J]. J Clin Immunol, 2020, 40(5): 671-81.
[3] ZHOU H, CLAPHAM D E. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development [J]. Proc Natl Acad Sci U S A, 2009, 106(37): 15750-5.
[4] BLOMMAERT E, PEANNE R, CHEREPANOVA N A, et al. Mutations in MAGT1 lead to a glycosylation disorder with a variable phenotype [J]. Proc Natl Acad Sci U S A, 2019, 116(20): 9865-70.
[5] BLOMMAERT E, CHEREPANOVA N A, STAELS F, et al. Lack of NKG2D in MAGT1-deficient patients is caused by hypoglycosylation [J]. Hum Genet, 2022.
[6] MATSUDA-LENNIKOV M, BIANCALANA M, ZOU J, et al. Magnesium transporter 1 (MAGT1) deficiency causes selective defects in N-linked glycosylation and expression of immune-response genes [J]. J Biol Chem, 2019, 294(37): 13638-56.
|

